Extraction and fractionation of oils
Application
Before liquid hydrocarbons present in a source rock or stained reservoir sample can be analysed in detail, they must be isolated from the rock matrix. Traditionally, extraction from finely milled samples uses an organic solvent system, and the organic extract is referred to as extractable organic matter (EOM), although other terms can be used (e.g. total organic extract, TOE).
EOM yield provides a guide to source rock quality and degree of reservoir saturation by migrated oil, assuming organic mud additives make negligible contributions. It may also be possible for oil to be obtained from core plugs by centrifugation.
The extract (or centrifuged oil) comprises polar and highly polymeric material as well as hydrocarbons. It is termed bitumen, and can be further analysed in exactly the same way as oil (which is also, technically, bitumen). It is conventionally thought of in terms of four basic components (SARA composition):
- Saturates (i.e. aliphatic hydrocarbons or alkanes)
- Aromatics (i.e. aromatic hydrocarbons)
- Resins (also termed polars – polymeric material containing NSO compounds)
- Asphaltenes (largest and most polar compounds)
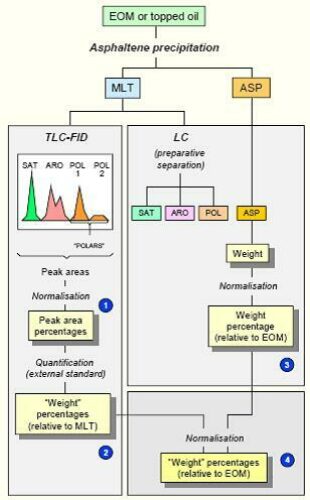
The relative abundances of the four bulk fractions can provide useful information about the origin of bitumen, including the presence of migrated oil and alteration processes, as well as contamination by organic drilling mud additives. This bulk compositional assessment is commonly referred to by the acronym SARA; it is usually determined by one of the following chromatographic methods that fractionates the remaining dissolved material (maltenes), following asphaltene precipitation:
- Iatroscan
- medium pressure liquid chromatography (MPLC)
Iatroscan is a TLC (thin layer chromatography) method, the purpose of which, in combination with asphaltene precipitation, is purely to determine the SARA composition, because it is a destructive technique (Karlsen & Larter 1991).
Alternatively, this quantitative information can be obtained by the non-destructive technique of MPLC. MPLC is also used in a semi-quantitative separation of saturates and aromatics for further analysis by GC and GC-MS, because it removes the most polar/polymeric compounds which adversely interfere with those chromatographic methods.
For some GC-MS analyses it can be necessary, or at least useful, to remove n-alkanes from the saturates fraction. For example, it is essential where distributions of C20+ acyclic isoprenoids are required, because of the unavoidable co-elution of some components with n-alkanes (unless biodegradation has selectively removed only n-alkanes).
It is useful where n-alkane abundance is relatively high to avoid chromatographic overloading when obtaining a reasonable signal from biomarkers, or possible signal suppression where co-elution with n-alkanes occurs on quadrupole instruments.
N-alkane isolation may be required for compound specific isotope analysis (CSIA).
Sample requirements
Bearing in mind the potential requirement for any subsequent GC, GC-MS or isotopic analyses, 100–200 g of unwashed cuttings (minimum of 50 g) or 20–30 g of washed cuttings, core or outcrop (5–10 g minimum) are required.
Samples are generally selected on the basis of representation of source rock or reservoir intervals, and often on the basis of TOC/Rock-Eval screening of source rocks. All rock samples require milling prior to extraction. Unwashed cuttings will require an initial washing, and a detergent based system is likely to be required where oil based mud is present.
Analytical procedure
Solvent extraction
Following any necessary washing of cuttings and milling of all rock samples, standard solvent extraction of milled source or reservoir rock samples, or drilling mud samples, is performed using an automated Soxtec Tecator instrument. Material is weighed accurately into pre-extracted thimbles and boiled for 1 h in approximately 80 mL of dichloromethane with 7% (vol/vol) methanol.
This is followed by 2 h of rinsing with the solvent. Elemental sulphur is removed by addition of copper blades (freshly activated in concentrated hydrochloric acid) to the extraction cups.
Quantification is performed by transferring an aliquot (10%) of the extract to a pre-weighed vial and evaporating to dryness. This aliquot is not used for subsequent analyses because of the unavoidable loss of volatiles.
For environmental samples (e.g. assessing surface seeps), extract yields are usually much lower than for source and reservoir rocks, and contamination can be a problem. Consequently, ultrasonic extraction is preferred to Soxtec.
The solvent system is the same, but no heating is involved during the ultrasonication (1 h). Sample work up is identical thereafter.
Deasphalting
The routine method is addition of excess pentane (40 x volume of oil/EOM) to the oil/extract (the latter has been evaporated almost to dryness before addition of a small amount of dichloromethane, ~3 x vol. EOM), following which the mixture is stored for at least 12 h in the dark before filtration or centrifuging. The weight of the dried asphaltenes is measured.
An extended (Moreoil) method can be applied, in which the weight of ~5 g oil is recorded and ~200 mL of n-pentane added (1:40 vol ratio oil:pentane). The mixture is agitated in an ultrasonic bath for 15 min and stored in the dark at room temperature for at least 24 h. The solution is then filtered using 0.45 mm paper and a water pump.
The filter cake is washed with n-pentane and the filter plus asphaltenes dried in an oven at 100°C for 30 min before being transferred to a desiccator and allowed to cool to constant weight.
Iatroscan
An Iatroscan MK-5 (TLC/FID) Analyser is normally used. 2 ml of extract or diluted oil is spotted on Chromarod S-III rods before elution with a sequence of solvents: hexane (25 min), toluene (8 min) and dichloromethane with 7% (vol/vol) methanol.
Each solvent is allowed to evaporate before the rods are placed into the next elution chamber. Before the rods are finally transferred to the analyser, they are heated for 90 s in a chamber at 60°C.
The relative abundance of saturates, aromatics and polars is obtained and combined with pre-determined asphaltene content to provide a SARA determination.
MPLC
The MPLC system is as described by Radke et al. (1980). It includes two HPLC (high performance liquid chromatography) pumps, sample injector and collector, IR and UV detectors, and two packed columns. The pre-column contains Kieselgel 100 (previously activated by heating at 600°C for 2 h) and the main column contains LiChroprep Si60 (previously activated at 120°C for 2 h under a helium flow to remove any water).
Approximately 30 mg of deasphaltened oil/EOM, diluted in 1 mL hexane, is injected and yields:
- Saturates – by elution with n-hexane
- Aromatics – by back-flushing the main column with hexane
- Polars – by back-flushing the pre-column with dichloromethane.
All fractions are concentrated to ~1 mL using a Turbovap, before transferring to small pre-weighed vials and reduced carefully to dryness before weighing.
The standard NGS SR-1 and/or NSG NSO-1 (both available from the Norwegian Petroleum Directorate) are analysed as the first and last sample on each sample set and checked against the acceptable range given in NIGOGA (Weiss et al. 2000).
N-alkane removal/isolation
Oil, EOM or saturates fraction (50 mg, or 150 mg for condensate), with internal standard added, in 7 mL dried toluene is added to 2 g of 5Å molecular sieve (previously activated by heating at 450°C for 16 h). The mixture is refluxed for 20 min. (followed by 40 min. cold rinse) under nitrogen using a Tecator Soxtec Avanti 2050 instrument.
After cooling the solution can be used for analysis of the branched/cyclic saturates.
For recovery of n-alkanes, the molecular sieve is filtered, using fresh toluene to wash the surface, and allowed to dry thoroughly. HF (6 mL of 30%) is added to break up the zeolite overnight, a process which is aided by ultrasonication after the initial reaction. N-alkanes are extracted by addition of pentane (0.5 mL) and elution from an inert micro-column, with further addition of pentane. The pentane is gently concentrated to 1 mL prior to further analysis of the n-alkanes.
Potential problems
The presence of organic based muds will affect EOM yields and SARA results: oil based mud (OBM) augments the saturates and polyglycols supplements the resins measurement. Where such contamination is suggested, confirmation may be obtained by GC analysis of the EOM (or oil).
Iatroscan and MPLC may not provide identical results, although there is no reason that well calibrated systems should not do so (Karlsen & Larter 1991). Problems may arise where data from different laboratories are compared, so it is recommended that the same method as used for most previous analyses is adopted. Data from Iatroscan are probably the more universally comparable.
Depending upon the concentration of n-alkanes, on rare occasions not all of them may be removed (the smallest are the most difficult to remove). In addition, all methods available for removal of n-alkanes have been found to result in depletion of other compounds with long n-alkyl chains, such as n-alkylcyclohexanes. So if GC-MS analysis of such compounds is required, a total saturates, rather than a branched/cyclics fraction, should be analysed.
References
Karlsen D.A., Larter S.R. (1991) Analysis of petroleum fractions by TLC-FID: applications to petroleum reservoir description. Organic Geochemistry 17, 603–617.
Radke M., Willsch H., Welte D.H. (1980) Preparative hydrocarbon group type determination by automated medium pressure liquid chromatography. Analytical Chemistry 52, 406−411.
Weiss H.M., Wilhelms A., Mills N., Scotchmer J., Hall P.B., Lind K., Brekke T. (2000) NIGOGA - The Norwegian Industry Guide to Organic Geochemical Analyses, Edition 4.0. 102pp. Available from http://www.npd.no/global/norsk/5-regelverk/tematiske-veiledninger/geochemical-analysis_e.pdf